多囊肾
多囊肾是常染色体显性遗传的遗传性疾病,父母如果发病,遗传到子女的几率是50%,多囊肾取决于两个因素:多囊肾基因、诱发基因激活的因素,两者同时存在才会导致发病。
多囊肾又叫Potter(Ⅰ)综合征、Perlmann综合征、囊胞肾等,它是一种常见的遗传性肾脏病,其病因是基因缺失,针对该病的治疗方式主要以预防为主,以下来具体了解下什么是多囊肾吧!
多囊肾的介绍
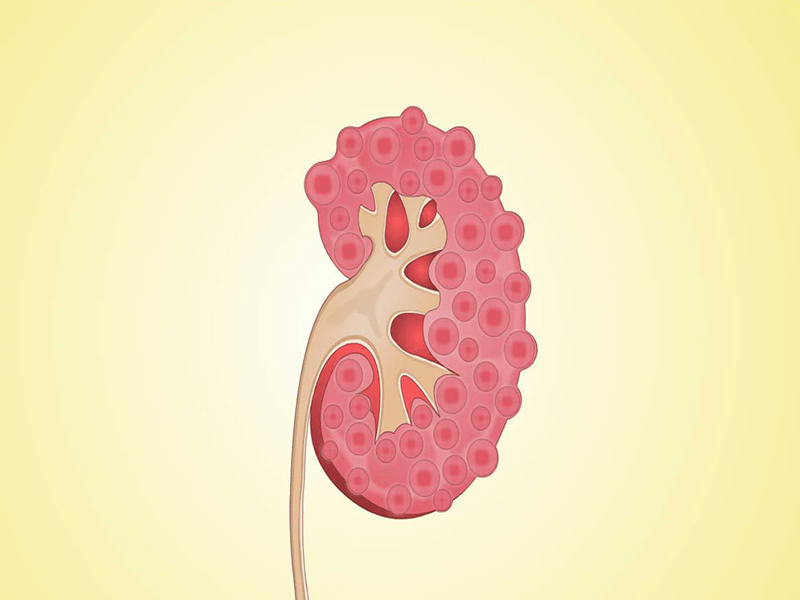
多囊肾病是一种影响肾脏和其他器官的疾病,肾脏上充满液体的囊群,称为囊肿,肾囊肿的体积大小和数量随着个体年龄增加而缓慢地增加,严重干扰了肾脏过滤血液中废物的能力。
囊肿的生长会导致肾脏不断变大,甚至导致肾衰竭,除了肾囊肿,囊肿也可能在其他器官发展,特别是肝脏。
多囊肾遗传规律
多囊肾的常染色体显性遗传型,其致病基因为PKD1;常染色体隐性遗传性多囊肾为PKHD1。
- 1. 男女发病几率相等;
- 2. 父母有一方患病,子女有50%获得囊肿基因而发病,如父母均患此病,子女发病率增加到75%;
- 3. 不患病的子女不携带囊肿基因,其下代(孙代)也不会发病,即不会隔代遗传,真正不发病前多无症状,当症状出现后,病情可发展很快。
多囊肾的临床症状
常见并发症包括高血压,背部或两侧疼痛,血尿,反复尿路感染,肾结石和心脏瓣膜异常。此外,患有多囊肾病的患者出现动脉瘤的风险会增加,常常出现在主动脉的大血管中或者大脑底部的血管中,动脉瘤破裂可能会危及生命。
多囊肾病有两种类型。常染色体隐性遗传型(婴儿型)多囊肾,发病于婴儿期,临床较罕见;常染色体显性遗传型(成年型)多囊肾,常于青中年时期被发现,也可在任何年龄发病。由于遗传基因的不同,进一步分为1型和2型。
临床表现
- 1. 肾肿大
- 2. 肾区疼痛
- 3. 血尿
- 4. 高血压
- 5. 肾功能不全
- 6. 多囊肝
- 7. 体格检查时可触及一侧或双侧肾脏,呈结节状
多囊肾病是一种相当常见的遗传疾病。其中常染色体显性遗传比常染色体隐性遗传更常见。常染色体显性遗传多囊肾病发病率为1/500-1000人,而常染色体隐性遗传发病率为1/2-4万人。
多囊肾的病因
多囊肾病主要是由于PKD1,PKD2和PKHD1基因突变所致。
PKD1或PKD2基因突变可导致常染色体显性遗传多囊肾病 。其中,PKD1基因突变可引起1型多囊肾病,而PKD2基因突变可引起2型多囊肾病。
研究表明,这些基因参与从细胞外传递化学信号到细胞核的过程。PKD1、PKD2基因所制备的蛋白质共同促进肾脏的正常发育功能。而PKD1或PKD2基因突变,可导致形成数千个囊肿,从而破坏了肾脏和其他器官的正常功能。
具有PKD2基因突变的人群,常见于女性,与PKD1基因突变患者相比,通常患病程度较轻,其症状如肾功能下降等往往在成年后出现。
多囊肾的治疗方法
本病尚无特异治疗方法,主要是控制血压和感染能有效延缓肾功能衰竭的进展,对不宜手术的病例给予对症治疗。
肾功能不全病人处理与慢性肾功能衰竭的治疗相同,肾绞痛发作可用各种镇痛药,并发感染时用抗生素治疗,常见的有抗血压治疗,抗尿路感染治疗,抗出血治疗,透析等。
多囊肾的生育干预
由于女性患者在病程早期并不妨碍妊娠及生育过程,但病程较晚则易并发高血压及孕毒症。
多囊肾属遗传病,患者的子女出生时携带致病基因的可能性为50%,因此早发现早干预,对于降低多囊肾的发生非常重要。多囊肾患者可以通过三代试管婴儿技术,在胚胎制备过程时,就能够及时剔除致病基因。
三代试管婴儿技术,在传统试管婴儿基础上,加入了单基因遗传病筛查。在胚胎移植前,通过基因检测,筛选出优质胚胎,确保婴儿不再患有基
因遗传病(包括多囊肾和其他基因遗传疾病),从而提升试管成功率。
尤其适用于伴侣双方中至少一人患有单基因遗传病(或为该类遗传病的携带者)以及已经生育过患有多囊肾孩子的家庭。
遗传疾病大全
| 罕见病汇总 | ||||
| 囊性纤维化 | 白化病 | 血友病 | 甲基丙二酸血症 | 唐氏综合症 |
| 特纳综合症 | 脆性X染色体综合症 | 克氏综合征 | 18三体综合征 | XXX综合征 |
| 帕陶氏综合症 | 马凡综合征 | 成骨不全症 | 镰刀型细胞贫血症 | 血红蛋白M病 |
| 地中海贫血 | 易位唐氏综合症 | 16号三体综合征 | 9号染色体三体 | 亨廷顿舞蹈症 |
| Alport综合征 | 鱼鳞病 | 结节性硬化症 | 苯丙酮尿症 | WAS综合征 |
| 大疱性表皮松解症 | 外胚层发育不良 | 枫糖尿病 | 多囊肾 | 先天性耳聋 |
| Bloom综合征 | Joubert综合征 | Laron综合征 | Lowe综合征 | 矮小症 |
| 胱氨酸贮积症 | 脊髓性肌萎缩症 | 瓜氨酸血症 | 卡氏综合征 | 高胱氨酸尿症 |
| 天使人综合症 | 维D缺乏性佝偻病 | 染色体脱染 | Meckel综合征 | bruton综合症 |
| 假肥大性肌营养不良 | 杜兴氏肌肉营养不良 | 软骨发育不全 | 色素性视网膜炎 | 先天性无指甲症 |
| 色盲 | α1抗胰蛋白酶缺乏症 | 埃莱尔-当洛综合征 | 先天性肌无力综合征 | 威廉姆斯综合征 |
| NBS断裂综合征 | 狐臭 | 共济失调 | 尼曼匹克病 | 肾上腺脑白质 |
| Sotos综合征 | 先天性夜盲症 | 异戊酸血症 | 镰刀型细胞贫血病 | 蚕豆病 |
| 先天性无虹膜 | 重症联合免疫缺陷病 | 先天性无/少汗症 | 先天性无痛无汗症 | 脊柱骨骺发育不全 |
| 鸟氨酸缺乏症 | 神经纤维瘤病 | 苯丙酮尿症 | 肝豆状核变性 | 先天性挛缩细长指 |